

In einer Studie haben Forscher der Universität Michigan einen neuen „Atlas“ des menschlichen Eierstocks vorgestellt, der die Fruchtbarkeitsbehandlung und die Hormonwiederherstellung revolutionieren könnte. Diese innovative Forschung bietet wertvolle Einblicke…

In einer Studie, die in der Zeitschrift Frontiers in Nutrition veröffentlicht wurde, haben Forscher die kausalen Zusammenhänge zwischen Darmmikroben und Schilddrüsenunterfunktion (Hypothyreose) aufgezeigt. Hypothyreose ist ein hormonelles Ungleichgewicht, das…

Bewegung als Heilmittel für Schlaflosigkeit
25.04.2024
Schlaflosigkeit, die durch Schwierigkeiten beim Einschlafen, beim Durchschlafen oder durch einen nicht erholsamen Schlaf gekennzeichnet ist, kann erhebliche Auswirkungen auf unsere allgemeine Gesundheit und unser Wohlbefinden haben. Jüngste Forschungsergebnisse…

Die Potenzmuskulatur und der Testosteronspiegel spielen eine entscheidende Rolle für die sexuelle Gesundheit und das Wohlbefinden von Männern. Eine starke Potenzmuskulatur ermöglicht eine bessere Kontrolle über die Erektion und…
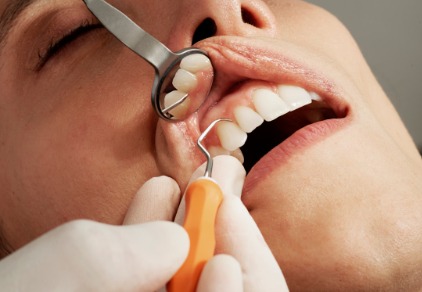
Amalgam: Wie schädlich ist die Zahnfüllung?
25.04.2024
Amalgamfüllungen sind in der Zahnmedizin weit verbreitet, aber sie sind auch umstritten. Der Grund dafür ist, dass Amalgam giftiges Quecksilber enthält. In diesem Artikel werden wir genauer untersuchen, wie…

Joghurt ist seit langem als nahrhaftes Lebensmittel bekannt, das verschiedene gesundheitliche Vorteile bietet. In den letzten Jahren hat das Interesse an der Rolle von Joghurt bei der Vorbeugung und…

Mit zunehmendem Alter steigt das Risiko, kognitive Beeinträchtigungen wie leichte kognitive Störungen (MCI) und Demenz zu entwickeln. Eine kürzlich durchgeführte Studie der Mailman School of Public Health der Columbia…

Das Reizdarmsyndrom (IBS) ist eine häufige Magen-Darm-Erkrankung, von der weltweite Millionen von Menschen betroffen sind. Es ist gekennzeichnet durch Symptome wie Bauchschmerzen, Blähungen und Völlegefühl, Durchfall und Verstopfung. Während…

In verschiedenen Studien wurde bestätigt, dass die Brunnenkresse mehr Vitamine enthält als jeder andere Kohl, jedes Kraut oder Ähnliches. Brunnenkresse, botanisch bekannt als Nasturtium officinale, ist eine krautige Pflanze…

Schuhe drinnen anlassen oder ausziehen?
22.04.2024
Die Frage, ob man seine Straßenschuhe in der Wohnung anlassen oder ausziehen sollte, ist ein Thema, das viele Menschen beschäftigt. Schließlich möchte man sein Zuhause sauber und hygienisch halten.…

In einer Welt, in der die geistige Leistungsfähigkeit geschätzt wird, suchen die Menschen ständig nach Möglichkeiten, ihre kognitiven Fähigkeiten zu verbessern. Von Gehirntrainingsübungen bis hin zu nootropischen Nahrungsergänzungsmitteln –…

Das neue Register für Organspenden
22.04.2024
Das neue Online-Organspenderegister ist ein Thema, das bei viele Bedenken und Skepsis hervorruft. Dennoch ist es eine wichtige Maßnahme, um die niedrige Anzahl an Organspendern zu erhöhen und somit…

In den letzten Jahren hat die weltweite Prävalenz von Typ-2-Diabetes mellitus (Typ-2-DiabetesM) und Prädiabetes stetig zugenommen. Angesichts der Millionen von Menschen, die mit diesen Erkrankungen leben, ist es von…

Eine medizinische Nährstoffanalyse am Menschen ist eine spezielle Untersuchung, die durchgeführt wird, um den Nährstoffstatus einer Person zu bewerten. Diese Art der Analyse ist besonders wichtig für Menschen mit…
Heilpraktiker: Eine kritische Betrachtung
20.04.2024
Alternative Therapieverfahren erfreuen sich zunehmender Beliebtheit, und immer mehr Menschen suchen einen Heilpraktiker auf. Doch warum ist das so? Und wie seriös sind diese Gesundheitsdienstleister eigentlich? Bundesgesundheitsminister Karl Lauterbach…

Viele Menschen suchen nach Möglichkeiten, ihren Körper und ihre Gesundheit optimal zu unterstützen. Leider gibt es jedoch auch schwarze Schafe, die versuchen, die gutgläubigen Verbraucher auszunutzen. Ein aktuelles Beispiel…

In der heutigen stressigen Welt ist es wichtiger denn je, sich um unsere geistige Gesundheit zu kümmern. Viele Menschen wenden sich dem Achtsamkeitstraining zu, um ihr allgemeines Wohlbefinden zu…

Das Altern wirkt sich auf verschiedene Aspekte unseres Körpers aus, auch auf unsere Muskeln. Wenn wir älter werden, werden unsere Muskeln allmählich schwächer, was sich auf unsere körperliche Kraft…
HÄUFIGE KRANKHEITEN, MEDIZINISCHE VERFAHREN UND GESUNDHEITSTIPPS

Erektionsstörung: Was hilft bei erektiler Dysfunktion?
Eine Erektionsstörung, auch erektile Dysfunktion genannt, ist ein häufiges Problem, das viele Männer betrifft. Besonders Männer über 60 Jahren sind…

Diabetiker zu zögerlich bei gesunder Ernährung und Bewegung
Typ-2-Diabetes ist eine chronische Erkrankung, die durch einen hohen Blutzuckerspiegel gekennzeichnet ist, weil der Körper nicht in der Lage ist,…

Augenringe: Ursachen und Behandlung
Augenringe sind ein häufiges Problem, das viele Menschen betrifft. Sie können sowohl ein kosmetisches als auch ein medizinisches Anliegen sein.…

Können Trockenfrüchte das Osteoarthritis-Risiko senken?
Osteoarthritis ist eine weit verbreitete degenerative Gelenkerkrankung, von der weltweit Millionen von Menschen betroffen sind. Sie ist gekennzeichnet durch Schmerzen,…

Neuer Entzündungsindex hilft bei Management von Autoimmunkrankheiten
Im Bereich der Autoimmunkrankheiten, bei denen sich das Immunsystem gegen den Körper wendet, den es eigentlich schützen soll, ist die Suche…

Können Psycho-Darmmikrobiotika bei Depressionen helfen?
Depressionen wirken sich nicht nur emotional auf den Einzelnen aus, sondern haben auch erhebliche Auswirkungen auf sein allgemeines Wohlbefinden, seine…

Reizdarmsyndrom: Neues Verfahren hilft Patientin nach 50 Jahren Durchfall und Bauchschmerzen
Wie stellt man fest, ob man ein Reizdarmsyndrom hat? Anhaltender Durchfall oder Verstopfung, starke Bauchschmerzen und ständige Abgeschlagenheit – wenn…

Diabetes: Beeinflussen Kartoffeln den Blutzuckerspiegel positiv oder negativ?
Sind Kartoffeln für Diabetiker geeignet? Kartoffeln stehen im Ruf, zur Gewichtszunahme beizutragen und das Risiko für Typ-2-Diabetes zu erhöhen. Deshalb…

Was tun bei Scharlach? – Ursachen und Behandlungen
Scharlach (Scarlatina) wird durch eine Infektion mit Bakterien verursacht, die Streptokokken A genannt werden. Es sind die gleichen Bakterien, die…

Forschung: Wirkung von Glycin und N-Acetylcystein auf den Alterungsprozess
ÜBERSICHT1 Lässt sich der Alterungsprozess umkehren?2 Den Alterungsprozess verstehen3 Glycin und N-Acetylcystein schützt vor oxidativem Stress4 GlyNAC vs. Glutathion5 Wirkung…

Kaffee und Diabetes: Welchen Einfluß Kaffee auf Diabetes und Gewicht (BMI) hat
Einfluss und Wirkung von Kaffee auf Diabetes Typ 2: Studien zeigen, dass Kaffee mit einem um etwa 25 Prozent verringerten…
Stimmbandentzündung – Ursachen, Symptome, Dauer, Ansteckungsgefahr, Behandlung
Was tun, was hilft bei einer Stimmbandentzündung? Was mit Husten und Heiserkeit (Dysphonie) beginnt, kann sich schnell zu einer Entzündung…

Borderline oder eher hochsensibel?
Viele Merkmale überschneiden sich zwischen der hochsensiblen Person (HSP) und der Borderline- Persönlichkeit. Eine hochsensible Person (HSP) und die Borderline-Persönlichkeit…

Studie: Cluster-Kopfschmerzen und ihre Auswirkungen auf die Leistungsfähigkeit
Auswirkungen und Folgen von Cluster-Kopfschmerzen Cluster-Kopfschmerzen sind kurze, aber extrem schmerzhafte Kopfschmerzattacken, die mehrere Tage oder unter Umständen sogar mehrere…
Gesundheit ist ein hohes Gut

Trotz der medizinischen Fortschritte der letzten Jahrzehnte ist eine Reihe von Krankheiten und Gesundheitsproblemen nach wie vor weltweit verbreitet. Nach Angaben von GlobalIssues.org sterben jährlich etwa 36 Millionen Menschen an nicht übertragbaren Krankheiten wie Krebs (Brustkrebs, Prostatakrebs, Lungenkrebs, Darmkrebs, etc.), Herz-Kreislauf-Erkrankungen (Herzinsuffizienz; Bluthochdruck, Atherosklerose, Herzrhythmusstörungen, Herzinfarkt, etc.), Diabetes mellitus (Zuckerkrankheit) sowie chronisch obstruktive Lungenerkrankungen ( COPD ) als auch Asthma bronchiale.
Unter den übertragbaren Krankheiten (Ansteckung, Infektion), sowohl viralen als auch bakteriellen Ursprungs, sind AIDS/HIV, Tuberkulose und Malaria die häufigsten und fordern jährlich Millionen von Todesopfern. Ein weiteres Gesundheitsproblem, das zum Tod führt oder zu anderen Krankheiten beiträgt, ist die Unterernährung, insbesondere bei Kindern, die am stärksten davon betroffen sind. Mehr als 7 Millionen Kinder unter 5 Jahren sterben an Unterernährung.
Ein weiteres weltweit verbreitetes Gesundheitsproblem sind Verletzungen durch Unfälle. Diese Verletzungen, darunter Knochenbrüche, Frakturen und Verbrennungen, können das Leben eines Menschen stark beeinträchtigen oder sogar tödliche Folgen haben. Dazu gehören auch Infektionen als Folge der Verletzung oder einer schweren Verletzung im Allgemeinen.
In vielen Fällen trägt auch der persönliche Lebensstil zu einem schlechten Gesundheitszustand bei. Dazu gehören Rauchen und ungesunde Ernährung. Das kann übermäßiges Essen oder Unterernährung sein. Bewegungs- und Schlafmangel, übermäßiger Alkoholkonsum, schlechte Mundhygiene sowie genetische Störungen sind weitere Faktoren, die die Gesundheit zum Teil erheblich beeinträchtigen und im schlimmsten Fall zum Tode führen können.
Obwohl die meisten dieser Krankheiten und Gesundheitsprobleme vermeidbar wären, ist einer der Hauptgründe für globale Krankheiten, dass etwa eine Milliarde Menschen keinen Zugang zu Gesundheitssystemen und qualitativ hochwertigen Medikamenten haben.
Psychische Gesundheit weltweit
 Die WHO umschreibt psychische Gesundheit als „einen Zustand des Wohlbefindens, in dem der Einzelne seine eigenen Fähigkeiten erkennt, mit den normalen Belastungen des Lebens umgehen kann, produktiv und erfolgreich arbeiten kann und einen Beitrag zu seiner Gemeinschaft leisten kann“.
Die WHO umschreibt psychische Gesundheit als „einen Zustand des Wohlbefindens, in dem der Einzelne seine eigenen Fähigkeiten erkennt, mit den normalen Belastungen des Lebens umgehen kann, produktiv und erfolgreich arbeiten kann und einen Beitrag zu seiner Gemeinschaft leisten kann“.
Psychische Gesundheit ist mehr als die Abwesenheit psychischer Störungen. Sie wird als das Ausmaß kognitiver, emotionaler und verhaltensbezogener Zustände beschrieben, die das soziale und emotionale Wohlbefinden, die Lebensqualität und die Produktivität einer Person beeinträchtigen. Eine psychische Erkrankung kann die geistige Leistungsfähigkeit, aber auch die körperliche Verfassung einer Person vorübergehend oder dauerhaft stark beeinträchtigen.
Psychische Erkrankungen sind in den USA, Kanada und vielen anderen hochentwickelten Industriestaaten eine der häufigsten Ursachen für gesundheitliche Beeinträchtigungen bis hin zu Arbeitsunfähigkeit, Berufsunfähigkeit, etc. Beispiele für solche psychischen Störungen sind zum Beispiel Schizophrenie, ADHS, Stimmungsstörung (DMDD), Depressionen, schwere Depression, bipolare Störungen, Angststörungen, posttraumatische Belastungsstörungen und Autismus.
Gesunde Ernährung und körperliche Bewegung

Ein wesentlicher Faktor für die Erhaltung der Gesundheit ist die Ernährung. Eine gesunde Ernährung besteht aus einer Vielzahl pflanzlicher und ausgewählter tierischer Lebensmittel, die den Körper mit den notwendigen Nährstoffen versorgen.
Diese Nährstoffe liefern dem Körper die notwendige Energie. Nährstoffe sind wichtig für den Aufbau und die Stärkung von Knochen, Muskeln und Sehnen und regulieren Körperprozesse. Und sauberes Trinkwasser ist wichtig für das Wachstum und die Gesundheit jedes Menschen.
Der menschliche Körper benötigt auch Makronährstoffe in relativ großen Mengen, darunter Proteine, Kohlenhydrate, Fette und Fettsäuren. Mikronährstoffe – Vitamine und Mineralstoffe – werden in relativ geringen Mengen aufgenommen, sind aber für die Körperprozesse unentbehrlich.
Die mediterrane Ernährung wird häufig mit gesundheitsfördernden Wirkungen in Verbindung gebracht, da sie einige bioaktive Verbindungen wie Phenolverbindungen, Isoprenoide und Alkaloide enthält.
Körperliche Aktivität verbessert oder erhält die körperliche Fitness, die allgemeine Gesundheit und wirkt sich positiv auf das Wohlbefinden aus. Sie stärkt die Muskeln und verbessert das Herz-Kreislauf-System. Laut den National Institutes of Health gibt es vier Arten von Bewegung: Ausdauer, Kraft, Beweglichkeit und Gleichgewicht.
Gesunder und erholsamer Schlaf

Erholsamer Schlaf ist ein wesentlicher Bestandteil der Gesundheit. Bei Kindern ist Schlaf darüber hinaus für Wachstum und Entwicklung von entscheidender Bedeutung. Schlafmangel ist mit einem erhöhten Risiko für eine Reihe von chronischen Krankheiten und Gesundheitsproblemen verbunden. Darüber hinaus gibt es Hinweise darauf, dass Schlafmangel sowohl mit einer erhöhten Anfälligkeit für Krankheiten als auch mit einer langsameren Genesung nach einer Krankheit einhergeht.
In einer Studie wurde festgestellt, dass Menschen mit chronischem Schlafmangel (sechs Stunden Schlaf pro Nacht oder weniger) viermal häufiger an einer Erkältung erkrankten als Menschen, die sieben Stunden oder mehr pro Nacht schliefen. Aufgrund der Rolle, die der Schlaf bei der Regulierung des Stoffwechsels spielt, kann unzureichender Schlaf auch eine Rolle bei der Gewichtszunahme oder umgekehrt bei der Gewichtsabnahme spielen.
Gesundheitsversorgung
Maßnahmen zur Verbesserung der Gesundheit, die auf Forschung und medizinisch-wissenschaftlich entwickelten Prinzipien und Verfahren beruhen, werden von ausgebildeten Fachkräften aus den Bereichen Medizin, Pflege, Ernährung, Pharmazie, Sozialarbeit, Psychologie, Ergotherapie, Physiotherapie und anderen Gesundheitsberufen durchgeführt.
Wellness- und Gesundheitsprogramme am Arbeitsplatz werden zunehmend von Unternehmen genutzt, um die Gesundheit und das Wohlbefinden ihrer Mitarbeiter zu verbessern und krankheitsbedingte Fehlzeiten zu reduzieren.
Welche Faktoren beeinflussen die persönliche Gesundheit?
Die persönliche Gesundheit jedes Menschen hängt zum Teil von den aktiven, passiven und unterstützten Signalen ab, die Menschen in Bezug auf ihre eigene Gesundheit wahrnehmen und entsprechend handeln.
Dazu gehören persönliche Maßnahmen zur Vorbeugung und/oder Begrenzung der Auswirkungen von Krankheiten. Dazu gehören Körperpflegepraktiken, die Infektionen und Krankheiten vorbeugen, wie Duschen, Baden und Händewaschen mit Seife, Zähneputzen und Zahnseide, die sichere Lagerung und Zubereitung von Lebensmitteln und vieles mehr.
Die persönliche Gesundheit hängt zum Teil auch von der sozialen Struktur eines Menschen ab. Die Aufrechterhaltung tragfähiger sozialer Beziehungen, ehrenamtliche Tätigkeiten und andere soziale Aktivitäten werden mit einer positiven psychischen Gesundheit und einer höheren Lebenserwartung in Verbindung gebracht.
Eine US-amerikanische Studie unter Senioren über 70 Jahren ergab, dass regelmäßige ehrenamtliche Tätigkeiten mit einem geringeren Sterberisiko verbunden waren als bei älteren Menschen, die keine ehrenamtlichen Tätigkeiten ausübten, unabhängig von ihrem körperlichen Gesundheitszustand. Eine weitere Studie aus Singapur ergab, dass ehrenamtlich tätige Rentner deutlich bessere kognitive Leistungen, weniger depressive Symptome, ein besseres psychisches Wohlbefinden und eine höhere Lebenszufriedenheit aufwiesen als nicht ehrenamtlich tätige Rentner.
Anhaltender psychischer Stress kann sich negativ auf die Gesundheit auswirken und ist ein Risikofaktor für kognitive Beeinträchtigungen und depressive Erkrankungen. Stressmanagement ist die Anwendung von Methoden, die entweder Stress reduzieren oder die Stresstoleranz erhöhen. Entspannungstechniken sind körperliche Methoden zum Abbau von Stress und Angst.
Zu den psychologischen Methoden gehören kognitive Therapie, Meditation und positives Denken, die durch eine Verringerung der Stressreaktion wirken. Durch die Verbesserung von Fertigkeiten wie Problemlösung und Zeitmanagement wird die Unsicherheit verringert und das Selbstvertrauen gestärkt, was wiederum die Reaktion auf Stresssituationen, in denen diese Fertigkeiten eingesetzt werden können, verringert.
Gesundheitsgefahren am Arbeitsplatz
 Neben den Sicherheitsrisiken bergen, weltweit betrachtet, viele Arbeitsplätze auch Risiken für arbeitsbedingte Erkrankungen und andere langfristige Gesundheitsprobleme. Zu den häufigsten Berufskrankheiten gehören verschiedene Formen der Migräne, Pneumokoniose, darunter Silikose und Kohlenarbeitspneumokoniose (schwarze Lungenkrankheit). Asthma ist eine weitere Atemwegserkrankung, für die viele Arbeitnehmer besonders anfällig sind. Die Beschäftigten können auch anfällig für Hautkrankheiten wie Ekzeme, Dermatitis, Urtikaria, Sonnenbrand und Hautkrebs sein. Andere Berufskrankheiten, die Anlass zur Sorge geben, sind das Karpaltunnelsyndrom und Bleivergiftungen.
Neben den Sicherheitsrisiken bergen, weltweit betrachtet, viele Arbeitsplätze auch Risiken für arbeitsbedingte Erkrankungen und andere langfristige Gesundheitsprobleme. Zu den häufigsten Berufskrankheiten gehören verschiedene Formen der Migräne, Pneumokoniose, darunter Silikose und Kohlenarbeitspneumokoniose (schwarze Lungenkrankheit). Asthma ist eine weitere Atemwegserkrankung, für die viele Arbeitnehmer besonders anfällig sind. Die Beschäftigten können auch anfällig für Hautkrankheiten wie Ekzeme, Dermatitis, Urtikaria, Sonnenbrand und Hautkrebs sein. Andere Berufskrankheiten, die Anlass zur Sorge geben, sind das Karpaltunnelsyndrom und Bleivergiftungen.
Mit dem Anstieg der Zahl der Arbeitsplätze im Dienstleistungssektor in den Industrieländern arbeiten immer mehr Menschen beispielsweise in sogenannten Bürojobs (in Deutschland sind es circa 18 Millionen Menschen), was in vielen Ländern völlig neue Probleme, wie die zunehmende Adipositasrate (Fettleibigkeit) und Probleme mit Stress am Arbeitsplatz und Überarbeitung zur Folge hat.
Medizin Doc: Weitere Beiträge & Gesundheitsnews der Medizin Doc Redaktion:
- Hypotonie: Blutdruck zu niedrig was tun, was hilft sofort? …
- Schwindelgefühl im Kopf – Was tun, was hilft? …
- Katzenallergie ignorieren? Welche Symptome treten auf bei einer Katzenallergie?…
- Was die Wissenschaft über die Wirksamkeit von Tai Chi und Qigong sagt …
- Soleus-Muskel-Pushups: Wie sich Stoffwechsel und Fettverbrennung im Sitzen ankurbeln läßt …
- Forschung: Wie gefährlich sind Antioxidationsmittel für die Gesundheit?
- Schmerzen und Verspannung: Solarplexus beruhigen, entspannen und Blockade lösen
- Vitamin D Tagesdosis: Wie viel ist zu viel?
- Ayurveda: Was bewirkt Haritaki – Welche Wirkung hat die Pflanze auf den Körper?
- Was tun gegen Blasenentzündung – was hilft?