Die Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia (ALSP) ist eine neurologische Erkrankung, die durch Veränderungen in bestimmten Bereichen des Gehirns gekennzeichnet ist.
ÜBERSICHT
Symptome und Krankheitsverlauf von ALSP
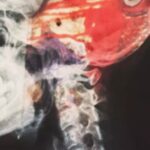
Ein charakteristisches Merkmal der Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia (Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia; ALSP) ist die Leukoenzephalopathie, die eine Veränderung einer Art von Hirngewebe, der so genannten weißen Substanz, darstellt.
- Die weiße Substanz besteht aus Nervenfasern (Axonen), die von einer Myelin genannten Substanz umhüllt sind, die sie isoliert und schützt.
Die Axone gehen von Nervenzellen (Neuronen) aus und leiten Nervenimpulse durch den ganzen Körper. Bereiche, in denen dieses Hirngewebe geschädigt ist (Läsionen der weißen Substanz), können mit der Magnetresonanztomographie (MRT) sichtbar gemacht werden.
Ein weiteres Anzeichen für die Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia (ALSP) sind Schwellungen, so genannte Sphäroide, in den Axonen des Gehirns, die auf eine Axonschädigung hinweisen.
- Ebenfalls häufig bei ALSP sind abnorm pigmentierte Gliazellen. Gliazellen sind spezialisierte Gehirnzellen, die die Neuronen schützen und erhalten.
Es wird angenommen, dass die Schädigung von Myelin und Neuronen zu vielen der neurologischen Anzeichen und Symptome bei Menschen mit ALSP beiträgt.
Die ersten Symptome von Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia (ALSP) treten in der Regel ab dem vierzigsten Lebensjahr auf und verschlimmern sich mit der Zeit.
Persönlichkeitsveränderungen, einschließlich Depressionen und Verlust sozialer Hemmungen, gehören zu den frühesten Symptomen von ALSP.
Bei den betroffenen Personen kann es zu Gedächtnisverlusten und zum Verlust der Exekutivfunktion kommen, das heißt der Fähigkeit, Handlungen zu planen und umzusetzen sowie Problemlösungsstrategien zu entwickeln.
Der Verlust dieser Funktion beeinträchtigt Fähigkeiten wie Impulskontrolle, Selbstkontrolle und angemessene Aufmerksamkeitslenkung. Einige Menschen mit Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia haben leichte Anfälle, meist nur zu Beginn der Erkrankung.
Mit dem Fortschreiten der ALSP kommt es zu einer schweren Beeinträchtigung des Denkens und der Argumentationsfähigkeit (Demenz).
Mit der Zeit werden auch die motorischen Fähigkeiten beeinträchtigt, und Menschen mit ALSP bekommen möglicherweise Schwierigkeiten beim Gehen.
Viele entwickeln ein Muster von Bewegungsanomalien, das als Parkinsonismus bezeichnet wird und ungewöhnlich langsame Bewegungen (Bradykinesie), unwillkürliches Zittern (Tremor) und Muskelsteifheit (Rigidität) umfasst.
- Das Muster der kognitiven und motorischen Probleme ist variabel, selbst bei Personen aus derselben Familie, wenngleich fast alle Betroffenen letztendlich nicht mehr in der Lage sind, zu gehen, zu sprechen und für sich selbst zu sorgen.
Früher wurde angenommen, dass es sich bei der Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia um zwei verschiedene Erkrankungen handelt, nämlich die hereditäre diffuse Leukoenzephalopathie mit Sphäroiden (HDLS) und die familiäre pigmentäre orthochromatische Leukodystrophie (POLD), die beide sehr ähnliche Schädigungen der weißen Substanz sowie kognitive und motorische Probleme verursachen.
Man vermutete zunächst, dass sich POLD durch das Vorhandensein von pigmentierten Gliazellen und das Fehlen von Sphäroiden unterscheidet; allerdings können auch Menschen mit HDLS pigmentierte Zellen und Menschen mit POLD Sphäroide haben. HDLS und POLD werden nun als Teil desselben Krankheitsspektrums betrachtet, für das Wissenschaftler und Wissenschaftlerinnen die Bezeichnung ALSP empfohlen haben.
Ursachen von ALSP
Die Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia wird durch Mutationen im CSF1R-Gen verursacht.
Dieses Gen gibt Anweisungen für die Herstellung eines Proteins, des so genannten Kolonie-stimulierenden Faktor-1-Rezeptors (CSF-1-Rezeptor), der sich in der äußeren Membran bestimmter Zelltypen, einschließlich Gliazellen, befindet.
- Der CSF-1-Rezeptor löst Signalwege aus, die viele wichtige zelluläre Prozesse steuern, wie Zellwachstum und -teilung (Proliferation) und die Reifung der Zelle zur Übernahme bestimmter Funktionen (Differenzierung).
CSF1R-Genmutationen bei ALSP führen zu einem veränderten CSF-1-Rezeptorprotein, das wahrscheinlich nicht in der Lage ist, Zellsignalwege zu stimulieren.
Es ist jedoch unklar, wie die Genmutationen zu Schäden an der weißen Substanz oder zu kognitiven und Bewegungsproblemen bei Menschen mit ALSP führen.
Vererbung von ALSP
Die Krankheit wird autosomal-dominant vererbt, was bedeutet, dass eine Kopie des veränderten Gens in jeder Zelle ausreicht, um die Störung zu verursachen.
In den meisten Fällen erbt eine betroffene Person die Mutation von einem betroffenen Elternteil. Andere Fälle sind auf neue Mutationen im Gen zurückzuführen und treten bei Menschen auf, in deren Familie die Krankheit nicht vorkommt.
Häufigkeit der Erkrankung
Obwohl die Prävalenz (Krankheitshäufigkeit, Anteil der erkrankten Personen an der Gesamtpopulation) unbekannt ist, gilt die Adulte Leukoenzephalopathie mit axonalen Sphäroiden und pigmentierten Glia als seltene Erkrankung.
- Da sie mit anderen Erkrankungen mit ähnlichen Symptomen verwechselt werden kann, wird ALSP möglicherweise unterdiagnostiziert.
VGT
Der Beitrag beschäftigt sich mit einem medizinischen Thema, einem Gesundheitsthema oder einem oder mehreren Krankheitsbildern. Dieser Artikel dient nicht der Selbst-Diagnose und ersetzt auch keine Diagnose durch einen Arzt oder Facharzt. Bitte lesen und beachten Sie hier auch den Hinweis zu Gesundheitsthemen!
Diagnose unbekannt: Leben mit einer seltenen Erkrankung
Quelle: Youtube/SPIEGEL TV; ARTE Re: