
Auf dem Röntgenbild erscheinen die Knochen so durchsichtig wie Glas. Doch für Betroffene der Glasknochenkrankheit ist die permanente Gefahr eines Knochenbruchs am schlimmsten. Dabei können noch viele weitere Probleme hinzukommen.
Osteogenesis imperfecta (OI) ist eine genetische oder vererbbare Krankheit, bei der die Knochen leicht brechen, oft ohne offensichtliche Ursache oder Verletzung. OI ist auch als Glasknochenkrankheit bekannt, und die Symptome können von leicht mit nur wenigen Frakturen bis schwer mit vielen medizinischen Komplikationen reichen.
Laut der Deutschen Gesellschaft für Osteogenesis imperfecta leiden bis sieben Menschen von 100.000 an der seltenen Erbkrankheit. Die Gesamtzahl der Betroffenen in Deutschland wird auf rund 6.000 Personen geschätzt.
ÜBERSICHT
- 1 Was passiert bei OI?
- 2 Wer erkrankt an Osteogenesis imperfecta?
- 3 Arten der Osteogenesis imperfecta
- 4 Symptome der Osteogenesis imperfecta
- 5 Ursachen der Osteogenesis imperfecta
- 6 Diagnose der Osteogenesis imperfecta
- 7 Behandlung der Osteogenesis imperfecta
- 8 Wer behandelt die Osteogenesis imperfecta?
- 9 Leben mit Osteogenesis Imperfecta
Was passiert bei OI?
Bei den meisten Menschen wird OI durch eine Veränderung oder einen Defekt in den Genen verursacht, die die Anweisungen für die Herstellung von Typ-I-Kollagen enthalten. Typ-I-Kollagen ist ein Material in den Knochen, das zu deren Festigkeit beiträgt. Der Gendefekt führt dazu, dass der Körper Kollagen falsch oder nicht in ausreichender Menge herstellt, was zu schwachen Knochen führt, die leichter brechen. Es gibt keine Möglichkeit, die Krankheit zu verhindern.
Wer erkrankt an Osteogenesis imperfecta?
Obwohl jeder mit OI geboren werden kann, haben Menschen mit einer familiären Vorgeschichte der Krankheit ein höheres Risiko, die Krankheit durch ein anormales Gen zu erben, das von einem oder beiden Elternteilen weitergegeben wird. Genetische Berater können Ihnen helfen, die Genetik der OI besser zu verstehen.
Arten der Osteogenesis imperfecta
Es gibt verschiedene Arten von OI, und je nach Schweregrad der Erkrankung oder der Art des zugrunde liegenden Gendefekts werden unterschiedliche Klassifizierungen verwendet. Typ I ist die mildeste und häufigste Form der OI. Typ II ist die schwerste Form der OI. Andere Formen der OI weisen Symptome auf, die zwischen Typ I und Typ II liegen. Im Folgenden finden Sie einen Überblick über die am häufigsten diagnostizierten Formen. Die übrigen Formen sind selten und werden noch erforscht.
Typ I
- Knochen, die bei leichten bis mittelschweren Traumata brechen können, wobei die meisten Knochenbrüche vor der Pubertät auftreten.
- Keine oder nur geringfügige Veränderungen der Statur mit zunehmendem Alter.
- Lockere Gelenke und Muskelschwäche.
- Blaue, violette oder graue Färbung der Sklera (des Augenweißes).
- Dreieckiges Gesicht.
- Gekrümmte Wirbelsäule mit der Gefahr einer Kompression der Wirbel (Wirbelknochen) mit zunehmendem Alter.
- Leichte oder keine Knochendeformität.
- Mögliche Veränderungen der Festigkeit und Farbe der Zähne.
- Möglicher Hörverlust.
- Normale Kollagenstruktur, aber weniger als die normale Menge.
Typ II
- Führt häufig zum Tod bei der Geburt oder kurz danach, weil die Atmung nicht mehr möglich ist.
- Zahlreiche Knochenbrüche, die vor der Geburt entstehen, während sich das Baby noch im Mutterleib befindet.
- Schwere Knochendeformierungen.
- Sehr kleine Statur.
- Unterentwickelte Lunge.
- Blaue, violette oder graue Färbung der Sklera.
- Unzureichend gebildetes Kollagen.
Typ III
- Der schwerste Typ unter denjenigen, die die Neugeborenenperiode überleben, und führt in der Regel zu den meisten körperlichen Behinderungen.
- Leicht gebrochene Knochen mit sehr wenig Trauma im Laufe des Lebens. (Knochenbrüche sind oft schon bei der Geburt vorhanden, und Röntgenaufnahmen können verheilte Knochenbrüche zeigen, die vor der Geburt entstanden sind).
- Kleine Statur.
- Blaue, violette oder graue Färbung der Sklera.
- Lose Verbindungen.
- Schlechte Muskelentwicklung in Armen und Beinen.
- Trommelförmiger Brustkorb.
- Dreieckiges Gesicht.
- Gekrümmte Wirbelsäule und Kompression oder Zusammenbruch der Wirbel.
- Mögliche Lungenprobleme, die sich mit zunehmendem Alter verschlimmern.
- Häufig schwere Knochendeformität.
- Mögliche Veränderungen der Festigkeit und Farbe der Zähne.
- Möglicher Hörverlust.
- Unzureichend gebildetes Kollagen.
Typ IV
- Knochen brechen leicht, manchmal schon vor der Geburt, wobei die meisten Knochenbrüche vor der Pubertät auftreten.
- Kleinere Statur als der Durchschnitt.
- Weiße oder blaue Färbung der Sklera.
- Leichte bis mäßige Knochendeformität.
- Wirbelkompression oder Zusammenbruch.
- Faßförmiger Brustkorb.
- Dreieckiges Gesicht.
- Mögliche Veränderungen der Festigkeit und Farbe der Zähne.
- Möglicher Hörverlust.
- Unzureichend gebildetes Kollagen.
Typ V
- Klinisch ähnelt die OI Typ IV in Aussehen und Symptomen.
- Ein dichtes Band, das auf Röntgenbildern an der Knorpelwachstumsplatte der langen Knochen zu sehen ist.
- Ungewöhnlich große Schwielen, so genannte hypertrophe Schwielen, an den Stellen von Frakturen oder chirurgischen Eingriffen. (Eine Schwiele ist ein Bereich neuen Knochens, der im Rahmen des Heilungsprozesses an der Frakturstelle gebildet wird).
- Verkalkung der Membran zwischen Speiche und Elle (den Knochen des Unterarms), die zu einer eingeschränkten Armbewegung führt.
- Möglicherweise lockere Verbindungen.
- Weiße Sklera.
- Keine Veränderungen an den Zähnen.
- „Maschenartiges“ Aussehen des Knochens bei Betrachtung unter dem Mikroskop.
- Veränderungen der Mineralien in den Knochen.
Typ VI
- Ähnelt in Aussehen und Symptomen der OI Typ IV.
- Die Diagnose wird nicht immer bei der Geburt gestellt, und die Symptome entwickeln sich im Laufe der Zeit.
- Unter dem Mikroskop sieht der Knochen wie ein Fischschuppen aus.
- Gebogene Wirbelsäule.
- Die Diagnose wird durch eine Knochenbiopsie oder genetische Untersuchungen gestellt.
- Veränderungen der Mineralien in den Knochen.
Typ VII
- Ähnelt in Aussehen und Symptomen der OI Typ II und Typ III.
- Weiße Sklera.
- Kleine Statur.
- Kurzer Humerus (Oberarmknochen) und kurzer Femur (Oberschenkelknochen).
- Möglicherweise kleinere Kopfgröße.
- Veränderungen im Prozess der Kollagenbildung.
Typ VIII
- Ähnelt in Aussehen und Symptomen der OI Typ II und Typ III.
- Weiße Sklera.
- Kleine Statur.
- Kurzer Humerus (Oberarmknochen) und kurzer Femur (Oberschenkelknochen).
- Möglicherweise kleinere Kopfgröße.
- Veränderungen im Prozess der Kollagenbildung.
Es gibt seltenere Formen der Krankheit, bei denen es sich im Allgemeinen um mittelschwere Formen handelt. Diese Formen ähneln den OI-Typen III oder IV. Einige dieser seltenen Formen beeinträchtigen nicht die Struktur des Kollagens, sondern die Funktion der knochenbildenden Zellen.
Symptome der Osteogenesis imperfecta
Alle Menschen mit OI haben schwache, brüchige Knochen. Manche Menschen mit OI haben im Laufe ihres Lebens nur wenige Knochenbrüche. Andere können im Laufe ihres Lebens Hunderte von Knochenbrüchen erleiden, darunter auch Knochenbrüche, die vor der Geburt auftreten.
Menschen mit OI können weitere Symptome haben, die von leicht bis schwer reichen und von Person zu Person variieren können. Dazu gehören:
- Missgebildete oder verbogene Röhrenknochen.
- Kleine Statur.
- Haut, die leicht blaue Flecken bekommt.
- Lose Verbindungen.
- Schwache Muskeln.
- Das Weiße der Augen (Sklera), das blau, violett oder grau aussieht.
- Ein Gesicht in Form eines Dreiecks.
- Ein Brustkorb in Form eines Fasses.
- Eine gebogene Wirbelsäule.
- Zusammenbruch oder Kompression der Wirbel in der Wirbelsäule.
- Brüchige, verformte oder verfärbte Zähne.
- Schwerhörigkeit.
- Ein verformtes Hüftgelenk, bei dem der Oberschenkelhals nach unten gebogen ist, ein Zustand, der Coxa vara genannt wird.
Ursachen der Osteogenesis imperfecta
Eine Mutation oder Veränderung in einem Gen verursacht OI. Gene enthalten Informationen, die bestimmen, welche Merkmale von den Eltern an den Menschen weitergegeben werden. Wir haben zwei Kopien der meisten unserer Gene, eine von jedem Elternteil.
Menschen mit OI haben ein Gen, das falsche Anweisungen für die Herstellung von Kollagen enthält, einer Substanz, die die Knochen stark macht. Das Gen führt dazu, dass der Körper nicht genügend Kollagen herstellt oder dass das Kollagen nicht richtig funktioniert. Dies führt zu schwachen Knochen, die leicht brechen.
Die meisten Menschen mit OI erben dieses Gen von einem Elternteil. Bei anderen Formen muss das Kind eine Mutation in einem Gen von beiden Elternteilen erben. Die Eltern müssen nicht unbedingt an OI erkrankt sein, um das Gen weiterzugeben, das die Krankheit verursacht. Manchmal wird das Gen von keinem Elternteil weitergegeben. Stattdessen hört das Gen von selbst auf, richtig zu funktionieren, bevor das Kind geboren wird.
Beherrschende OI
Die meisten Menschen mit OI haben eine dominante Form. Das heißt, sie erben eine normale Kopie und eine Kopie des Gens, das OI verursacht. Die abnorme Kopie des Gens ist stärker oder „dominant“ gegenüber der normalen Kopie des Gens. Dies führt dazu, dass eine Person OI hat. Eine Person mit einer dominanten Mutation hat eine 50-prozentige Chance (1 zu 2), die Störung an jedes ihrer Kinder weiterzugeben. Einige Kinder mit der dominanten Form der OI erben ein Gen, das OI verursacht, von einem Elternteil. Andere wiederum werden mit der dominanten Form der OI geboren, obwohl es keine familiäre Vorgeschichte der Erkrankung gibt und die Mutation in ihrem Genom zum ersten Mal in der Familie auftritt.
Rezessive OI
Einige Menschen mit OI haben eine rezessive Form der Krankheit. Menschen mit rezessiver OI haben Eltern, die nicht an OI leiden, aber beide ein abnormales Gen haben, das die Krankheit verursacht. Wenn beide Eltern das rezessive Gen für OI tragen, besteht eine 25 %ige Chance (1 von 4) pro Schwangerschaft, ein Kind mit der Krankheit zu bekommen. Nicht betroffene oder asymptomatische Geschwister einer Person mit rezessiver OI haben eine Zwei-Drittel-Chance (2 von 3), ein abnormales Gen zu tragen, das OI verursacht, und sind somit Träger der Krankheit. Wenn ein Elternteil aufgrund einer rezessiven Mutation an OI erkrankt ist, werden alle seine Kinder ein abnormales Gen tragen, das OI verursacht, aber nicht unbedingt an OI erkranken.
Diagnose der Osteogenesis imperfecta
Ärzte können OI folgendermaßen diagnostizieren:
- Nach der Familien- und Krankengeschichte fragen.
- Durchführung einer körperlichen Untersuchung.
- Bestellung von Röntgenaufnahmen und Knochendichtetests.
Darüber hinaus können Ärzte OI diagnostizieren und die Art der OI mit einem genetischen Bluttest feststellen, der die Veränderung des vererbten Gens aufspürt. Mit diesen Tests kann die OI bei den meisten Menschen, die sie haben, nachgewiesen werden. Manchmal können zusätzliche Gentests erforderlich sein. Menschen, die sich einem Gentest unterziehen, sollten einen Spezialisten oder genetischen Berater aufsuchen, der ihnen hilft, die Testergebnisse zu verstehen.
Behandlung der Osteogenesis imperfecta
Es gibt keine Heilung für OI. Das Ziel der Behandlung besteht je nach Art der OI darin, die Symptome zu verhindern oder zu kontrollieren, die Knochenmasse und die Muskelkraft zu erhöhen und die Fähigkeit der Betroffenen, unabhängig zu sein, zu maximieren. Diese Behandlungen umfassen:
Physikalische oder ergotherapeutische Behandlung
Menschen mit OI können von einer Physio- oder Ergotherapie profitieren, die den Betroffenen helfen kann:
- Aufbau von Muskelkraft, Verbesserung der Gelenkbewegungen, der Mobilität und der Grobmotorik sowie Vorbeugung von Knochenbrüchen.
- Lernen Sie, wie Sie Verletzungen vermeiden können.
- Sichere Durchführung von Aktivitäten des täglichen Lebens.
- Erholen Sie sich von Knochenbrüchen.
Therapeuten und Ärzte können auch Schwimmen zur Konditionierung und zum Kraftaufbau empfehlen.
Medikamente
Obwohl es keine zugelassenen Arzneimittel zur Behandlung der OI gibt, kann Ihr Arzt eine Therapie empfehlen, die für eine verwandte Erkrankung zugelassen ist. Ihr Arzt kann Ihnen etwas verschreiben:
- Knochenstärkende Medikamente, die zur Behandlung anderer Knochenerkrankungen zugelassen sind, können dazu beitragen, den Knochenabbau zu verlangsamen und die Häufigkeit und Schwere von Knochenbrüchen zu verringern.
- Schmerzmittel zur Behandlung von Schmerzen, die durch Knochenbrüche und chronische Knochenschmerzen verursacht werden.
Darüber hinaus werden derzeit einige Arzneimittel zur Vorbeugung von Komplikationen oder zur Behandlung von OI bei Erwachsenen und Kindern untersucht. Sprechen Sie mit Ihrem Hausarzt oder dem Kinderarzt Ihres Kindes über die Verwendung dieser Arzneimittel oder die Teilnahme an Studien.
Knochenpflege
Ein Orthopäde kann gebrochene Knochen mit einem Gips, einer Schiene oder einem Korsett behandeln. Manchmal ist eine Operation erforderlich, um einen gebrochenen Knochen zu reparieren.
Darüber hinaus führen die Ärzte Operationen durch, um gekrümmte oder verbogene Knochen, einschließlich der Wirbelsäule, zu stützen oder zu korrigieren. Viele Kinder mit OI unterziehen sich einer Stabeingriff, bei dem ein Metallstab in einen Knochen eingesetzt wird. Die Stabeingriffe werden durchgeführt, um den Knochen zu stützen und zu verhindern, dass der Knochen bricht. Einige dieser Stäbe sind „teleskopierbar“ und können so angepasst werden, dass sie sich verlängern, wenn ein Kind mit OI wächst.
Mobilitätshilfsmittel
Die Verwendung einer Mobilitätshilfe kann Menschen dabei helfen, tägliche Aktivitäten sicher auszuführen und Verletzungen zu vermeiden. Zu diesen Hilfsmitteln gehören:
- Stöcke und Krücken.
- Zahnspangen oder Prothesen.
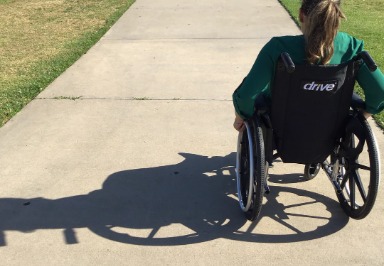
- Rollstühle.
Mund- und Zahnpflege
Einige Menschen mit OI haben:
- Zähne, die leicht splittern oder brechen.
- Veränderungen der Zahnfarbe und -form.
- Kleine Kiefergröße.
- Falsche Stellung der Zähne.
Regelmäßige zahnärztliche Untersuchungen und Pflege sind wichtig, um Zahnerkrankungen vorzubeugen und Biss, Ausrichtung und Aussehen der Zähne zu verbessern. Darüber hinaus müssen einige Menschen sehen:
- Mund-, Kiefer- und Gesichtschirurg, der sich auf Mund-, Kiefer- und Gesichtschirurgie spezialisiert hat.
- Kieferorthopäde, der die Zahnstellung und Kieferposition behandelt.
Audiologische Untersuchungen
Ärzte empfehlen Hörtests ab dem Kindesalter und regelmäßige Untersuchungen während des gesamten Lebens der Betroffenen. Es ist wichtig, einen Audiologen aufzusuchen, der auf die Betreuung von Menschen mit OI spezialisiert ist. Die Behandlung kann Folgendes umfassen:
- Hörgeräte, kleine elektronische Geräte, die außerhalb des Ohrs getragen werden und den Ton lauter machen.
- Cochlea-Implantate, kleine elektronische Geräte, die aus zwei Teilen bestehen, einem außen hinter dem Ohr und einem weiteren unter der Haut.
- Bei der so genannten Stapedektomie setzt der Chirurg eine Prothese oder ein künstliches Gerät in das Mittelohr ein, damit die Schallwellen das Innenohr erreichen können.
Wer behandelt die Osteogenesis imperfecta?
Menschen mit OI benötigen in der Regel ein Gesundheitsteam aus mehreren Ärzten und Gesundheitsdienstleistern. Zu Ihrem Gesundheitsteam können gehören:
- Allgemeinmediziner, die Erwachsene diagnostizieren und behandeln.
- Kinderärzte, die Kinder diagnostizieren und behandeln.
- Klinische Genetiker, die Kinder und Erwachsene mit genetischen Störungen diagnostizieren und behandeln.
- Orthopäden, die Knochen- und Gelenkerkrankungen behandeln und operieren und Erfahrung in der Behandlung von Menschen mit OI haben.
- Ergotherapeuten, die lehren, wie man Aktivitäten des täglichen Lebens sicher ausführt.
- Physiotherapeuten, die lehren, wie man Muskeln stärkt, sich von Knochenbrüchen erholt und Knochenbrüchen vorbeugt.
- Zahnärzte wie Kieferorthopäden und Mund-, Kiefer- und Gesichtschirurgen.
Leben mit Osteogenesis Imperfecta
Bestimmte Aktivitäten können Menschen mit OI helfen, gesund zu bleiben und Knochenbrüche zu vermeiden.
- Achten Sie auf eine nährstoffreiche Ernährung.
- Bewegen Sie sich so viel wie möglich. Regelmäßige körperliche Betätigung kann zur Stärkung von Muskeln und Knochen beitragen. Schwimmen und Wassergymnastik sind für Menschen mit OI eine gute Wahl, da bei Übungen im Wasser das Risiko von Knochenbrüchen gering ist. Sprechen Sie mit Ihrem Arzt oder Physiotherapeuten, um geeignete und sichere Übungen zu besprechen.
- Halten Sie ein gesundes Gewicht. Übergewicht erhöht das Risiko für viele Gesundheitsprobleme, wie Diabetes und Herzkrankheiten. Zusätzliches Gewicht belastet auch die Knochen, was für Menschen mit OI besonders ungesund ist.
- Rauchen Sie nicht, und vermeiden Sie Passivrauchen, denn auch Rauchen kann die Knochen schwächen.
- Trinken Sie nicht zu viel Alkohol oder Koffein, da dies Ihre Knochen schwächen kann.
- Suchen Sie eine Beratung auf oder sprechen Sie mit einer medizinischen Fachkraft, wenn Sie sich deprimiert oder ängstlich wegen der OI und ihrer Symptome fühlen.
Dieser Beitrag beschäftigt sich mit einem medizinischen Thema, einem Gesundheitsthema oder einem oder mehreren Krankheitsbildern. Dieser Artikel dient nicht der Selbst-Diagnose und ersetzt auch keine Diagnose durch einen Arzt. Bitte lesen und beachten Sie hier auch den Hinweis zu Gesundheitsthemen! Quellen: Der Beitrag basiert u.a. auf Informationen von MedlinePlus und Wikipedia lizenziert nach CC-by-sa-3.0 oder Open Government v3.0.




